


2020年から小学校でのプログラミング教育の必修化が開始されています。
小さな子供を持つママ達の間では、小学校や中学校でのプログラミング教育への疑問点や不安がどんどん大きくなっているのを感じます。

そこで、質問です。
あなたは、プログラミング必修化やロボット教室などについてどれくらい知っていますか?

あなたはこんな疑問ありませんか?
- ロボット教室ってなにするの?
- どうやって選べば良いのかよくわからないわ?
- いつから通わせたらいいの?
- おすすめのロボット教室を知りたいわ!
なんて方は、ロボット教室を徹底調査!した当サイトを参考にしてください。
当サイトでは、子供にロボット教室に通わせようと考えている親御さんのために、
などについて詳しく解説しています。

初めてのプログラミング教育には「ロボット」を使うのが1番です。
子どもの将来のために、「何か準備をしたい、新しいことに興味を持ってもらいたい」と思う方には最適な教室です!
2025年大学入試科目にプログラミングが追加!今後は避けては通れない

2020年の教育改革。
プログラミングに関する社会の動きは、小学生だけではなく、中学校、高校にまで広がっています。
プログラミング教育が小学校3年生から必修化されることはご存知の方も多いはず。
中でも注目すべきなのが、2025年1月に実施される大学入学共通テストの科目に『情報』を加えるということです。
安倍首相が議長の未来投資会議では、『AI時代の人材育成』という議題が話し合われました。
安倍首相はこの会議で、プログラミングなどに関する「情報科目」を、国語や英語と並ぶ基礎的科目として大学入試に追加する方針を表明ました。
【参考:未来投資会議】
安部元首相は、「プログラミングは現代の『読み・書き・そろばん』になる」という言葉も述べています。
どうりで、子供の将来のために、小学生の頃からプログラミングに触れさせたい親御さんが増えているわけです!
プログラミング教育 学校以外でも学習する人は増加中
プログラミング学習に対する親御さんの注目度は、年々高まってきています。
その表れとして、プログラミング教室は、最近の「子どもにさせたい習い事ランキング」などのアンケート調査でも上位にランクインしています。
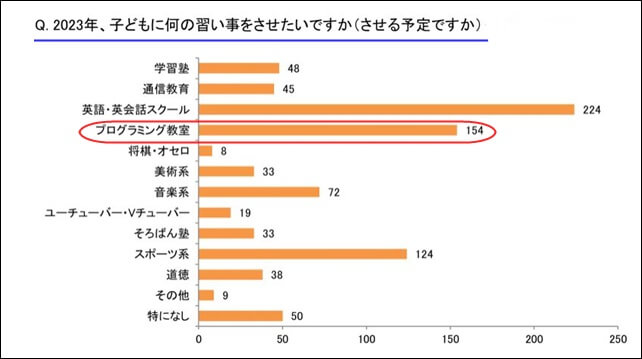
【引用:PR TIMES】
毎年年末に行っているイーラーニング研究所の「年末年始の習い事アンケート」の結果によると、
2023年にさせたい習い事として、「英会話スクール」についで「プログラミング教室」が2位になっています。
ロボット教室は今大注目の習い事!五感で感じるから身につきやすい!

小学生のプログラミング教育の目的は、難しいプログラミング言語を学ぶのではなく、「プログラミング的思考」を身につけることにあります。
つまり、問題を解決するためにはどうしたらいいのかを、論理的に順序立てて考える思考力を身に着けさせることが目的です。
でも、学校での教育は学校や地域ごとに差があるのが現状。
子供のプログラミング学習にロボット教室がBESTな理由

2020年のプログラミング必修化に対応するために、小学生にプログラミングを学ばせようと考えているなら、ロボット教室がおすすめです。
その理由としては、
理由1
理由2
ロボット教室の方がおすすめなのは、ロボットが動くのを五感で理解しながらプログラムを理解するから。
つまり、自分が組んだプログラミングで、ロボットを思い通りに動かせるのが、具体的でわかりやすいからです。
あなたの子供がまだ小学生ならなおさらです。
▼ロボット教室とプログラミング教室の違いは?通うならどっちがおすすめ?
では、さらに詳しく解説しています。

小学生におすすめロボット教室BEST4!これだけは要チェック!
 子供向けのプログラミング教室の主流は、ロボット教室といえます。
子供向けのプログラミング教室の主流は、ロボット教室といえます。
同じロボット教室でも、先生の質や授業内容などは様々です。
私は、息子と一緒に行けるところは無料体験に行き、実際に各教室の雰囲気や授業内容を体験してきました。
まずは、自分が通える範囲にある教室の無料体験に行ってみましょう。
個別カリキュラムが人気!LITALICOワンダー

LITALICO(リタリコ)ワンダーは、決まったカリキュラムがなく、子供の個性に合わせて授業内容を組み立ててくれるのが魅力。
少々料金は高めですが、内容を考えれば納得です。
ゲームやアプリをプログラミングするコース、ロボットプログラミングコース(2つ)、3Dプリンターを使った造形コースがあります。
東京・神奈川・埼玉・千葉にお住まいの方にはおすすめの教室。

オンライン授業もあるので、全国どこからでも受講できます!
無料体験もオンラインでOK!!
ここをチェック!
- 対象年齢:年長~
- 月謝:1回7,425円~(教室orオンライン、回数によって異なる)
- 教育用レゴを使用(SPIKEベーシック、SPIKE プライムセット)
- 教室数:24(東京、神奈川、埼玉、千葉のみ)
- 一部のコースでオンライン授業あり!
- 無料体験あり(オンラインが人気!)
ワンランク上のロボット教室!クレファス

クレファスは、人気のレゴを使ったロボットプログラミングが学べる教室。
小1からロボットプログラミングが学べるだけでなく、プレゼンテーションにも力を入れています。
教室内の大会はもちろん、レゴの世界大会への出場者を多数排出。
ここをチェック!
- 対象年齢:小1~(年長もあるが、プログラミングなし)
- 月謝:12,100円~
- 教育用レゴを使用(WeDo2.0、SPIKEプライム)
- 教室数:約120
- オンライン授業あり
- 無料体験あり
料金を抑えたい!近くに教室がない!
そんな方にはクレファスのオンライン講座「イークレファス」がおすすめ!

e-crefus(イークレファス)は、ロボットを自分で考えて組み立てる自習型のオンライン学習スタイル。テキストやロボット教材は実際の教室と同じものを使用しています。
自分のペースで学べる自習型学習でありながら、聞きたいこと、分からないこと、見てもらいたいものがあったら、待機中の先生とパソコン等からリモートで会話ができるので安心!
プログラミング重視のアーテックエジソンアカデミー

アーテックエジソンアカデミーは、学校で採用多数の教材メーカが開発したロボットを使用。
3年間のプログラムでじっくりとロボットプログラミングを学ぶことができます。
小3以下の子には、「自考力キッズ![]() 」という別カリキュラムがあり、年少からプログラミングを使ったロボット作りができます。
」という別カリキュラムがあり、年少からプログラミングを使ったロボット作りができます。
ここをチェック!
- 対象年齢:小3~(小2以下用のカリキュラムあり)
- 月謝:約10,000円
- 独自開発のアーテックロボットを使用
- 教室数:900以上
- 無料体験あり
ヒューマンアカデミーロボット教室

ヒューマンアカデミーは、全国に1600以上もの教室数を誇る最大規模のロボット教室。
他のロボット教室と比べてロボット教材、月謝ともに安く、コストパフォーマンスに優れたロボット教室と言えます。
通いやすさ、料金設定、授業内容、どれをとっても満足できるレベル。
実際に通わせている保護者の声はとても参考になりますよね!
ここをチェック!
- 対象年齢:年長~
- 月謝:10,340円
- オリジナルのロボット教材やテキストは、日本を代表するクリエイターが監修。
- 教室数:1600以上
- 無料体験あり
プログラミングだけの教室もあります。
自宅がロボット教室に?!教室よりもリーズナブルさがうけてます!
近くに教室がない方でも大丈夫!
上記で紹介したLitalicoワンダーや ![]() イークレファスも自宅でできるコースもありますが、それ以外におすすめの2つを紹介します。
イークレファスも自宅でできるコースもありますが、それ以外におすすめの2つを紹介します。
Z会プログラミング講座

「KOOV]というロボットキットをつかった講座で、女の子でも楽しめるようなポップでカラフルなブロックを使います。
Z会オリジナルのテキストを使いながら自宅で好きな時に学習することができます(^^♪
アフレル

教育用レゴの「SPIKEプライム 」をテキスト付で販売。
セットアップも難しくなく、オリジナルのワークブックも充実した内容。
8才以上が対象でコスパがいいのが魅力。
こちら以外にも自宅でできるロボットプログラミング学習についてまとめた記事はこちら!
ロボット教材を使わないプログラミング学習(ゲーム作成など)に興味がある方はこちら!
自宅でゲーム作成を通してプログラミング学習をやりたい!
そんな方におすすめの記事はこちら。

【個別指導or動画】子供向けオンラインロボット・プログラミング教室12選!
ロボット教室を選ぶときに気を付けるべきポイント

子供をロボット教室に通わせようと思っているけど、どうやって選んだらいいのかわからない人が多いと思います。
- 料金
- 通いやすさ(家からの距離)
- 授業内容(カリキュラム)
- 実際に通っている人の口コミ
なかでも、重要なのが「料金」と「通いやすさ」です。
また、値段だけ安くても、肝心のカリキュラムや先生の質が悪くては行かせる意味がなくなってしまいます。
ロボット教室の料金はいくらかかるの?相場は?

ロボット教室で必要となる料金には、「入会金」「月謝」「ロボットキット代」の3つが必要になります。
それぞれおおよその相場はこのくらい。
- 入会金:1万円前後
- 月謝:約1万円~2万円
- ロボット代:2万円代~6万円(※教室ごとに使うロボットキットが違う)
ほとんどのロボット教室がロボットを購入する形になっています。
教室によっては、進級時に追加キットの購入があったり、教室で貸出してくれるところもあります。
ロボット教室選び関する悩みはこちらの記事で解決!


ロボット教室選びは、1つの項目だけでなく、総合的に判断する必要があります。
なので、詳しいロボット教室の選び方の参考に、
当サイトの別記事「子供向けおすすめロボット教室6選!失敗しない選び方」をご覧ください。
▼ママ目線で徹底比較!▼
プログラミング学習は何歳から始めたらいいの?実は小学生がはじめ時!

ロボット教室に通わせるベストなタイミングはずばり、小学生です。
幼稚園児でも通える教室もありますが、プログラミングをやっているところは限られてきます。
小学1年生から通い始めるご家庭は多く、各教室でも低学年用のコースが用意されています。
小学生3年生以上になると、より各教室でのコースの内容は充実してきます。
高度なロボットプログラミングも行うようになり、ますます子供の能力が開花していくことが期待されます。
我が子が通える教室があるのかわからない方は必見です!

ロボット教室って何をするの?

まだロボット教室についてリサーチを始めたばかりの親御さんは、
「ところで、ロボット教室ってなにをするの?」という疑問を持っていると思います。
ロボット教室でのメインの授業内容はこちら。
- ブロックを使ってロボットを作成
- 作ったロボットをプログラミングで思い通りに動かす
教室によっては、理科や物理の内容をおり交ぜて、もののしくみを解説する授業も行っています。

ロボット教室で身につく能力は?

ロボット教室に通うことで、身につく能力はいくつかあります。
例えば、「プログラミング的思考」「創造力」「空間認識力」「発想力」「集中力」などが身につくとされています。
子供の頃にプログラミングの考え方や仕組みを学ばせることで、将来的に、様々な職業への準備ができることにもなります。
▼こちらも参考にしてください!
-

ロボット教室はどんな効果があるの?1年通って感じた息子の嬉しい変化とは?
「ロボットを作ることが、子どもにとってどんな役に立つの?」 「ただ、遊んでいるようにしか見えない」 なんて、ロボット教室 ...
続きを見る

最後にもう一度!行っておくべき無料体験教室BEST4!
ロボットを使ったプログラミング教室であれば、どこでも同じというわけではありません。
教室によって授業の進め方や取り組む内容が異なります。
幸いなことに、ほとんどのロボット教室が無料体験を行っています!
ここは外せない!行っておくべき無料体験教室BEST4!
一覧表で特徴をまとめました。
| 子供ごとに個別のカリキュラムでの授業。直営店だけなので先生の質は間違いなし。オンラインでの受講 も人気! |
| 本格的なロボットプログラミングが学べる点は魅力的。大会出場に力を入れている。 オンライン講座の【e-crefus】イークレファス |
| ロボット制作よりもプログラミング重視の教室。教室数も生徒数も急増中! |
| 全国に1600以上もの教室数を誇る、最大手ロボット教室。 |

ロボット教室選び成功のカギは、体験教室をうまく活用すること!

ロボット教室はいろいろありますが、HPだけ見ていても違いはよくわかりませんよね。
そんな時は、無料体験を利用しましょう。
「もっとしっかり考えて選べばよかった」
なんて後悔することにもなりかねません。
なので、できるだけ早い段階から、「どこのロボット教室がいいのかな~」と情報を集めることをおすすめします。
子供が行ってくれるのであれば、面倒に思わずに、2、3回は体験に行ってみてください。
実際に教室にいくことで、不安な点や疑問点がクリアになりますので、無料体験は必須です。
知識や経験が増えてくると、教室ごとの違いがよくわかるようになります。
親が自分の目で見て感じた感覚で、子供に合いそうなロボット教室を選んであげることが、一番確実な選び方かなと思います。
ぜひとも、体験教室を上手に活用してみてくださいね。